ISSN: 2708-552X RNPS: 2495
Presentación de caso
Síndrome de Ehlers-Danlos tipo cifoescoliótico
Ehlers-Danlos syndrome kyphoscoliotic type
Leander José Aguilar Ricardo 1 https://orcid.org/0000-0001-8696-4262
Juan Carlos Pérez Almaguer 1 https://orcid.org/0009-0005-9515-0347
Liana Rosa Reyes Leyva 1 https://orcid.org/0009-0003-5104-5356
Lilibeth Pérez González 1 https://orcid.org/0000-0001-9042-9414
María Claudia Cruz García 1 https://orcid.org/0009-0008-5659-0018
1Facultad de Ciencias Médicas Mariana Grajales Coello. Universidad de Ciencias Médicas de Holguín, Cuba.
*Autor para la correspondencia. Correo electrónico: leander010918@gmail.com
Recibido: 18/12/2024
Aprobado: 06/01/2025
RESUMEN
El síndrome de Ehlers-Danlos es un grupo de trastornos hereditarios del tejido conectivo; clínico y genéticamente heterogéneo, caracterizado por la síntesis anormal de colágeno que afecta piel, ligamentos, articulaciones, vasos sanguíneos y otros órganos. Se reconocen seis subtipos principales, entre los cuales se incluye el cifoescoliótico, caracterizado por hipotonía muscular grave al nacer, hipermovilidad articular, luxaciones, cifoescoliosis grave progresiva, hiperelasticidad cutánea, hábito marfanoide, osteopenia, ojos frágiles y fragilidad vascular. Se realiza este informe con el objetivo de caracterizar un caso clínico de sospecha de síndrome de Ehlers-Danlos tipo cifoescoliótico. Se trata de un paciente que acude al servicio de Medicina Interna por presentar dolor y pérdida de fuerza en la región del bíceps braquial, al cual se le constató la presencia de varias signos clínicos de los descritos anteriormente. Se concluye que por su infrecuencia está enfermedad suele ser subdiagnosticada por lo que se hace necesario divulgar sus características clínicas.
Palabras clave: cutis elástica; enfermedad del colágeno; síndrome de Ehlers-Danlos
ABSTRACT
Ehlers-Danlos syndrome is a clinically and genetically heterogeneous group of inherited connective tissue disorders characterized by abnormal collagen synthesis affecting skin, ligaments, joints, blood vessels, and other organs. Six main subtypes are recognized, including kyphoscoliotic, characterized by severe muscular hypotonia at birth, joint hypermobility, dislocations, severe progressive kyphoscoliosis, skin hyperelasticity, marfanoid habitus, osteopenia, fragile eyes and vascular fragility. This report is made with the objective of characterizing a clinical case of suspected Ehlers-Danlos syndrome, kyphoscoliotic type. This is a patient who came to the Internal Medicine service for presenting pain and loss of strength in the biceps brachii region, which on physical examination revealed the presence of various clinical signs of those described above. It is concluded that due to its infrequency, this disease is usually underdiagnosed, so it is necessary to disclose its clinical characteristics.
Keywords: collagen disease; Ehlers-Danlos syndrome; elastic skin
Introducción
El síndrome de Ehlers-Danlos (Ehlers-Danlos syndrome; EDS, por sus siglas en inglés) es un grupo de trastornos hereditarios del tejido conectivo, clínica y genéticamente heterogéneos, caracterizados por hiperextensibilidad cutánea, pobre cicatrización de heridas, hipermovilidad articular y friabilidad de los tejidos.(1)
Es caracterizado por la síntesis anormal de colágeno que afecta piel, ligamentos, articulaciones, vasos sanguíneos y otros órganos, causado por mutaciones en los genes que codifican el colágeno fibrilar tipo I, III y V o enzimas comprometidas en la modificación post-translacional de dichos colágenos. Se reconoce la existencia de 6 subtipos principales: clásico, hiperlaxitud, vascular, cifoescoliosis, artrocalasia y dermatoparaxis.(2)
Entre estos subtipos se incluye el cifoescoliótico, caracterizado por hipotonía muscular grave al nacer, hipermovilidad articular, luxaciones, cifoescoliosis grave progresiva, hiperelasticidad cutánea, hábito marfanoide, osteopenia, ojos frágiles asociados con la ruptura del globo ocular y, ocasionalmente, fragilidad vascular.(1)
Esta variante es causada por la deficiencia de procolágeno-lisina 2-oxa-glutarato 5-dioxigenasa 1 (PLOD1), producida por una mutación en homocigosis o heterocigosis compuesta de los alelos PLOD1.(1)
La primera descripción del síndrome data de 1682 y se la debemos a Job Von Meekeren (1611-1666) un cirujano de Ámsterdam. El siguiente informe apareció en Moscú en 1891, donde el Dr. A. N. Tschernogobow presentó dos casos (un niño de 17 años y una mujer de 50).(2)
La aportación más importante de este autor fue que asoció las múltiples manifestaciones de estas personas, y dijo que eran debidas a un trastorno generalizado del tejido conectivo. A él corresponde el mérito de la descripción clásica del síndrome que en realidad debería conocerse por su nombre.(2)
Posteriormente en París (1899) Edward Ehlers presentó otro caso en una reunión de la Sociedad de Veneorología y Dermatología. El enfermo presentaba hiperlaxitud articular y variados y múltiples problemas ortopédicos. Tenía la piel hiperextensible y había desarrollado lesiones pigmentadas sobre las prominencias óseas debido a traumatismos mínimos y también presentaba debido a los mismos una tendencia hemorrágica relativamente importante.(2)
Nueve años más tarde, en 1908 Henri Alexandre Danlos, expuso otro caso. Afirmó que las lesiones sobre las prominencias óseas eran postraumáticas y que el paciente tenía un defecto inherente, que él denominó "cutis laxa". FrederichParkes-Weber en un artículo publicado en el Diario de la Sociedad Británica de Dermatología propuso el nombre de síndrome de ElhersDanlos para denominar a esta enfermedad. Este nombre ganó aceptación entre los miembros.(2)
La incidencia de síndrome de Ehlers-Danlos se estima de 1:5000, siendo la mayor prevalencia la del tipo con hipermovilidad que afecta a uno de cada 10000 a 15000 individuos, seguido de los tipos clásico y vascular.(3)
Por lo general, tener articulaciones excesivamente laxas, la piel frágil o quebradiza y antecedentes familiares de síndrome de Ehlers-Danlos basta para hacer el diagnóstico. En formas menos comunes de este síndrome, realizar pruebas genéticas sobre muestras de sangre puede ayudar a confirmar el diagnóstico y a descartar otros problemas. Para el subtipo hipermóvil, la forma más común, no existen pruebas genéticas disponibles.(2)
En este artículo se describen las características del cuadro clínico de un caso de sospecha de síndrome de Ehlers-Danlos, acontecido en el Hospital Militar de Holguín "Fermín Valdés Domínguez" en el año 2023.
Presentación de caso
Se trata de un paciente masculino de 18 años de edad, de raza blanca, procedencia rural, que actualmente se encuentra cumpliendo con el servicio militar activo, por lo que es soldado. Acude a consulta por pérdida de fuerza y dolor en la región del brazo izquierdo.
Posee antecedentes patológicos personales de asma bronquial y escoliosis, para los cuales no lleva tratamiento. Sufrió una fractura en la muñeca izquierda hace aproximadamente 7 años. No refiere operaciones, transfusiones ni reacción alérgica a medicamentos. Como antecedentes patológicos familiares refiere que su padre padece de asma bronquial y su abuela materna de diabetes mellitus. No presenta hábitos tóxicos, solo bebe alcohol en ocasiones especiales.
Fue ingresado en el servicio de Medicina Interna del Hospital Militar de Holguín, pues desde hacía aproximadamente un mes comenzó a presentar pérdida de fuerza en el brazo izquierdo, haciéndole difícil la flexión del antebrazo al cargar peso, refiere que "trabaja descargando baños" y que desde hace un mes "se le hace imposible cargar cubos con ese brazo".
Posteriormente comenzó a presentar dolor en la cara anterior del brazo izquierdo, que no se irradiaba a ninguna otra zona, de carácter pulsátil, de intensidad moderada, que sólo aliviaba con la administración de calmantes, presentándolo diariamente, principalmente en horario nocturno o cuando está en reposo, presentándolo hasta que logra conciliar el sueño.
Al examen físico se constató: hábito marfanoide (paciente longilíneo con extremidades superiores alargadas); a la inspección en la actitud de pie se puede observar cifoescoliosis en la región dorsal (figura 1); hiperlaxitud de todas sus articulaciones, más marcada en la articulación del codo izquierdo, la cual sobrepasa los 30º más allá del eje longitudinal del brazo (figura 2); presencia de pectusexcavatum (figura 3).
Con respecto al miembro doloroso, a la inspección se observa disminución del tono del bíceps braquial izquierdo, el cual no fue doloroso a la palpación ni a la movilización pasiva, con marcada pérdida de fuerza a la flexión, con fuerza a la extensión conservada.
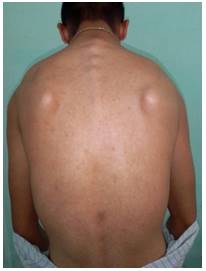
Figura 1. Escoliosis dorsal visible a simple vista

Figura 2. Hiperlaxitud de la articulación del codo izquierdo

Figura 3. Pectusexcavatum Se indican los complementarios siguientes: Hemoglobina: 140 g/L; leucograma: global 10x109/L, neutrófilos 0.51, linfocitos 0.41, monocitos 0.03, eosinófilos 0.05; eritrosedimentación: 4 mm/h; TGO: 16 UI/L; TGP: 23 UI/L; GGT: 39 UI/L; glicemia: 4.0 mmol/L; colesterol: 3.5 mmol/L; triacilglicéridos 80 mmol/L. Se indican radiografía de hombro, no patológica, y de tórax, que confirma la escoliosis.
Se indica tratamiento con: dipirona (500 mg), 1 tab/8h si dolor; Vitamina B12 (1000 microgramos), 1 cc IM/día. Responde adecuadamente al tratamiento.
Discusión
El síndrome de Ehlers-Danlos tipo cifoscoliótico (kEDS por sus siglas en inglés) es un raro trastorno autosómico recesivo del tejido conjuntivo caracterizado por cifoescoliosis progresiva, hipotonía muscular congénita, hiperextensibilidad cutánea grave y marcada hipermovilidad y dislocación.(4,5)
Además, la fragilidad de la piel con cicatrización anormal, hábito marfanoide, hernia, microcórnea, dismorfología facial, esclerótica azul, errores de refracción, osteopenia y ruptura arterial podría también presentarse en pacientes con kSED.(4,5)
Esta enfermedad presenta una serie de criterios que permiten hacer su diagnóstico positivo (la presencia de 3 o más criterios mayores en la infancia es diagnóstica). Signos mayores: Laxitud articular generalizada, hipotonía muscular intensa desde el nacimiento, escoliosis progresiva con inicio en el nacimiento, fragilidad escleral y rotura del globo ocular. Signos menores: Fragilidad tisular, magulladuras fáciles, rotura arterial, hábito marfanoide, microcórnea y osteopenia.(2)
En el caso que se presenta en este informe, el paciente presenta algunas de las características antes descritas, tales como el hábito marfanoide, la hiperlaxitud articular, la cifoescoliosis progresiva y signos de hipotonía, elementos que hacen sospechar que padece esta enfermedad, y por las que se recomienda hacer seguimiento por el servicio de Genética Médica para realizar un diagnóstico más certero.
Algunos de los diagnósticos diferenciales de esta patología son: el síndrome de Marfan, el síndrome de Loeys-Dietz, el síndrome de X frágil, la osteogénesis imperfecta, el síndrome de Stickler, el síndrome de Williams y la acondroplasia.
El diagnóstico del síndrome de Ehlers-Danlos es limitado ya que en la actualidad no hay ninguna prueba sencilla, objetiva y fiable que permita diagnosticar todos y cada uno de sus tipos. Muchas personas con la enfermedad requieren un diagnóstico clínico confirmado habitualmente por un genetista especializado en enfermedades raras.(2)
Especialistas como dermatólogos, pediatras y reumatólogos también pueden diagnosticar el trastorno. Para diagnosticarlo correctamente los profesionales deben valorar los antecedentes familiares y la historia clínica del enfermo. Como fue señalado, la mayor parte de los enfermos tienen una mezcla de síntomas de todas las categorías y no entran en una categoría claramente definida.(2)
La severidad de las manifestaciones clínicas puede variar incluso entre los miembros de la misma familia, e incluso cada persona de la misma familia puede estar afectada de manera diferente.(2)
Debido a esta dificultad para el diagnóstico, al hecho de que se considera una enfermedad rara y a que la hiperlaxitud articular es un signo clínico que muchas veces por obvio los profesionales de la salud no lo tienen en cuenta, es que el síndrome de Ehlers-Danlos es una enfermedad que a menudo pasa desapercibida siendo en la actualidad subdiagnosticada.(2)
La hiperlaxitud articular se valora según diversas escalas. La Sociedad Internacional de Síndrome de Ehlers-Danlos determinó en el año 2017 la utilización correcta del Beighton score como el procedimiento más adecuado para establecer el diagnóstico de hiperlaxitud articular generalizada y, además, presentó una actualizada clasificación del síndrome de Ehlers-Danlos, describiendo 13 tipos de EDS y sus respectivos criterios clínicos.(7,8)
Tofts et al.7 establecieron estos criterios, proporcionando una escala de evaluación que es la más utilizada hoy en día y es considerada aún el pilar fundamental de los criterios que se han propuesto hasta ahora.(7,8)
La escala de Beighton otorga al paciente 1 punto por cada una de las siguientes características: extensión pasiva de la quinta metacarpofalange que sobrepase los 90 grados, aposición pasiva del pulgar al antebrazo, hiperextensión del codo de más de 10 grados, hiperextensión de la rodilla de más de 10 grados y flexión del tronco que permita que las palmas de la mano se posen en el suelo.(2,8)
En dichos criterios se puntualiza que cada extremidad de los cuatro primeros es un item por separado según fuera la derecha o la izquierda, generando una posible puntuación de 9. (2,8,9)
Ruiz-Botero et al.1 publicaron en 2019 un caso de una paciente adolescente colombiana con kEDS, la cual al nacer presentó hipotonía generalizada, luxación congénita de cadera bilateral y perforación gástrica neonatal, así como cifoscoliosis progresiva fue percibida a los 2 años de edad y que requirió una operación quirúrgica a los 10 y a los 15 años, pectumexcavatum, luxación del tobillo, índice de Beighton 9/9, entre otras manifestaciones.(1)
Malfait et al.9 reportaron el caso de un hombre de 42 años de origen norteamericano, nacido de padres no consanguíneos, con un fenotipo que incluía cifoscoliosis grave, enfermedad pulmonar restrictiva, baja talla, hipoacusia leve, reducción de la masa muscular y hallazgos vasculares, como disección de la arteria celíaca a los 41 años y oclusión completa de la arteria mesentérica superior con flujo compensatorio mediante una mesentérica inferior agrandada y tortuosa.(1,9)
En 2020, Fairweather et al.4 reportaron un caso de un paciente de 17 años que presentaba hipotonía muscular congénita, cifoescoliosis progresiva, hiperextensibilidad y fragilidad severas de la piel, marcada hipermovilidad articular y dislocación, dismorfologías faciales, esclera azul, errores de la refracción y osteopenia. Al compararse con otros casos, conocemos que el descrito en este informe no alcanza un alto grado manifestaciones clínicas como en otros casos de mayor gravedad.(4)
A la hora de indicar el tratamiento, se debe tener en cuenta en qué fase de la vida se encuentra el paciente, ya que los síntomas varían según las mismas, de tal forma que algunos autores han decidido dividir el avance de la enfermedad en 3 fases progresivas, correspondiendo cada una a una década de vida.(9,10)
El tratamiento ha de centrarse en la promoción de estilos de vida saludables y ejercicios individualizados, la fisioterapia y la psicoterapia. Puede conseguirse mejorar la estabilidad mediante ejercicios de baja resistencia para aumentar el tono muscular, y durante la fase aguda de la enfermedad es necesario el reposo de la articulación afectada.(10)
Por lo general se requiere aplicación de calor o frío y a veces el uso de férulas, son útiles los masajes, ultrasonido y ultratermia y se recomienda paracetamol o antiinflamatorios por períodos cortos.(2,10)
Conclusiones
El síndrome de Ehlers-Danlos es una enfermedad heterogénea, con diversas manifestaciones clínicas que permiten clasificarlas en 6 principales subtipos, entre los que se encuentra el tipo cifoescoliótico. El mismo presenta entre otras características la presencia de cifoescoliosis progresiva, hiperlaxitud articular, hábito marfanoide e hipotonía muscular generalizada, características todas presentes en el caso descrito. Dada la infrecuencia de la enfermedad queda normalmente subdiagnosticada, por lo que se hace necesario conocer los signos clínicos para poder reconocerla en los pacientes que lleguen a consulta médica.
Referencias bibliográficas
1. Ruiz Botero F, Ramírez Montañoa D, Pachajoa H. Síndrome de Ehlers-Danlos cifoescoliótico-FKBP14 en una paciente adolescente: primer reporte de caso colombiano. Arch Argent Pediatr.2019;117(3):e274-e278. Puerto Martinez Marianela.
2. Puerto Martínez M. Caracteizacion clínia y manejo del Síndrome de Ehlers Danlos. Rev Ciencias Médicas.2017[citado 19/05/2023];21(4):124-150. Disponible en:
http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1561-31942017000400018&lng=es.
3. Alonso Valle A, Huerta Pérez G, Ramírez Santos I, Candelaria Gómez B. Síndrome de Ehlers-Danlos, a propósito de un caso. Jornada de residentes de genética clínica; 2021/03/01-05.La Habana: Centro Nacional de Genética Médica;2021.Disponible en:
https://www.google.com/url?sa=t&source=web&rct=j&url=https://instituciones.sld.cu/cngm/files/2021/03/pdf-10.pdf&ved=2ahUKEwiJldCHrNH-AhWAD1kFHZeaBtQQFnoECBQQAQ&usg=AOvVaw3k8Bh5NUAEw0hSAkGUlTAY
4. Fairweather D, Bruno KA, Darakjian AA, Bruce BK, Gehin JM, Kotha A, et al. High overlap in patients diagnosed with hypermobile Ehlers-Danlos syndrome or hypermobile spectrum disorders with fibromyalgia and 40 self-reported symptoms and comorbidities. Front Med.2023 [citado 08/05/2023];10:1096180. Disponible en:
https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2023.1096180/full
5. Ni X, Jin C, Jiang Y, Wang O, Li M, Xing X, et al. The first case report of Kyphoscoliotic Ehlers-Danlos syndrome of chinese origin with a novel PLOD1 gene mutation. BMC Med Genet.2020 [citado 30/03/2023]; 21(1):214. Disponible en:
https://bmcmedgenet.biomedcentral.com/articles/10.1186/s12881-020-01154-3
6. Ritelli M, Chiarelli N, Cinquina V, Vezzoli M, Venturini M, Colombi M. Looking back and beyond the 2017 diagnostic criteria for hypermobile Ehlers-Danlos syndrome: A retrospective cross-sectional study from an Italian reference center. Am J Med Genet A .2024 [citado 20 /08/2024];194(2):174-194. Disponible en: https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63426
7. Tofts LJ, Simmonds J, Schwartz SB, Richheimer RM, O'Connor C, Elias E, et al. Pediatric joint hypermobility: a diagnostic framework and narrative review. Orphanet J Dis Raras.2023 [citado 20/09/ 2024];18(1):104. Disponible en:
https://ojrd.biomedcentral.com/articles/10.1186/s13023-023-02717-2
8. González Adonis F, Bratz J, Sandoval Ramírez M, Guerrero Nancuante C. Hipermovilidad articular y síndrome de Ehlers-Danlos: consideraciones desde el cuidado en enfermería. Iatreia.2019[citado 02/05/2023];32(4):346-353. Disponible en:
https://revistas.udea.edu.co/index.php/iatreia/article/view/335144
9. Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, et al. The 2017 international classification of the Ehlers–Danlos syndromes. Am J Med Genet C Semin Med Genet.2017 [citado 02/05/2023];175(1):8-26. Disponible en: https://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31552
10. Virág Ralovich F, Kiss N, Horváth K, Kárpáti S, Medvecz M. Up-to-date classification and multidisciplinary symptoms of Ehlers-Danlos syndromes. Orv Hetil.2019 [citado 03/05/2023];160(16):603-612. Disponible en: https://akjournals.com/view/journals/650/160/16/article-p603.xml
Conflicto de intereses
Los autores declaran que no existe conflicto de intereses.
Financiación
No se recibió financiación para la realización del presente artículo.
Contribución de autoría
LJAR: Conceptualización, Metodología, Investigación, Administración del proyecto, Supervisión, Redacción-borrador original, Revisión y Redacción
JCPA: Supervisión, Redacción-borrador original, Revisión y Redacción
LRRL: Supervisión, Redacción-borrador original, Revisión y Redacción
LPG: Supervisión, Redacción-borrador original, Revisión y Redacción
MCCG: Supervisión, Redacción-borrador original, Revisión y Redacción
Publicación cuatrimestral de la Universidad de Ciencias
Médicas de Holguín ![]() CC-BY-NC 4.0
CC-BY-NC 4.0